
Scleroderma isn't just a skin condition. It’s a silent, progressive disease that turns the body’s own repair system against it. Instead of healing, the body overproduces collagen-turning soft tissue into hard, tight bands. This isn’t just about wrinkled fingers. It’s about lungs that can’t expand, hearts that struggle to pump, and digestive tracts that refuse to move food. And it’s happening to more people than most realize.
What Exactly Is Scleroderma?
Scleroderma is a rare autoimmune disease where the immune system attacks connective tissue, causing excessive collagen buildup that hardens skin and internal organs. Also called systemic sclerosis, it’s not contagious, not inherited in a simple way, and not caused by lifestyle. It’s a malfunction deep inside the immune system, and we still don’t fully know why it happens.
There are two main types. Localized scleroderma (or morphea) only affects the skin-patches of thickened, discolored skin that might look like bruises or scars. It rarely spreads. The real danger lies in systemic scleroderma, which can hit the lungs, heart, kidneys, and gut. About 90% of people with systemic scleroderma develop problems in their digestive system. Eight in ten develop lung scarring. One in three face heart complications.
Women are four times more likely to get it than men. Most people are diagnosed between 30 and 50. It’s rare in children and older adults, though that’s changing as people live longer with the disease.
Early Warning Signs You Can’t Ignore
Many people live with scleroderma for years before they get a diagnosis. Why? Because the first signs look like other things. Cold fingers? That’s just Raynaud’s. Fatigue? Must be stress. Trouble swallowing? Maybe heartburn.
But here’s the pattern: Raynaud’s phenomenon shows up in 90% of cases-often 5 to 10 years before anything else. Fingers turn white, then blue, then red when cold or stressed. It’s not just sensitivity to cold. It’s a sign that tiny blood vessels are spasmming and dying. If you’ve had Raynaud’s for more than a few years and now your fingertips feel tight or shiny, that’s a red flag.
Another early sign is sclerodactyly. Your fingers start to feel stiff. You can’t bend them fully. The skin looks tight, like it’s been stretched over a balloon. You might notice your knuckles don’t move the same way. This happens in 95% of systemic scleroderma cases.
Antinuclear antibodies (ANA) show up in 95% of patients. That’s not a diagnosis on its own-many healthy people have ANA-but when it shows up with Raynaud’s and skin changes, it’s a major clue.
How It’s Diagnosed-And Why It Takes So Long
The average patient sees 3.2 doctors over 18 months before getting the right diagnosis. That’s because symptoms overlap with lupus, rheumatoid arthritis, and even chronic fatigue syndrome.
Doctors look for three things: skin changes, internal organ involvement, and specific antibodies. Blood tests check for anti-Scl-70 (linked to lung scarring), anti-centromere (linked to limited disease), and anti-RNA polymerase III (linked to rapid skin thickening and higher cancer risk).
Imaging helps too. A high-resolution CT scan catches lung fibrosis early. An echocardiogram checks for pulmonary arterial hypertension-something that kills 30-40% of scleroderma patients. A skin biopsy isn’t always needed, but if your doctor suspects deep tissue involvement, they might order one.
The key is catching it before organs are damaged. That’s why specialists now recommend that anyone with long-standing Raynaud’s get screened, even if they don’t have obvious skin changes.
Different From Other Autoimmune Diseases
Scleroderma doesn’t look like lupus. Lupus flares with rashes, joint pain, and kidney issues-but it doesn’t cause hard, tight skin. Rheumatoid arthritis attacks joints with inflammation. Scleroderma doesn’t cause swollen joints-it causes stiff ones because the skin around them is pulling tight.
Polymyositis weakens muscles. Scleroderma doesn’t. Mixed connective tissue disease shares symptoms but has a unique antibody (anti-U1 RNP) that scleroderma patients don’t have.
The biggest difference? Fibrosis. Scleroderma’s signature is excessive collagen-the stuff that makes skin firm. In other diseases, it’s inflammation that’s the problem. In scleroderma, the inflammation triggers a repair process that never stops. That’s why treatments for lupus or RA often don’t work here.
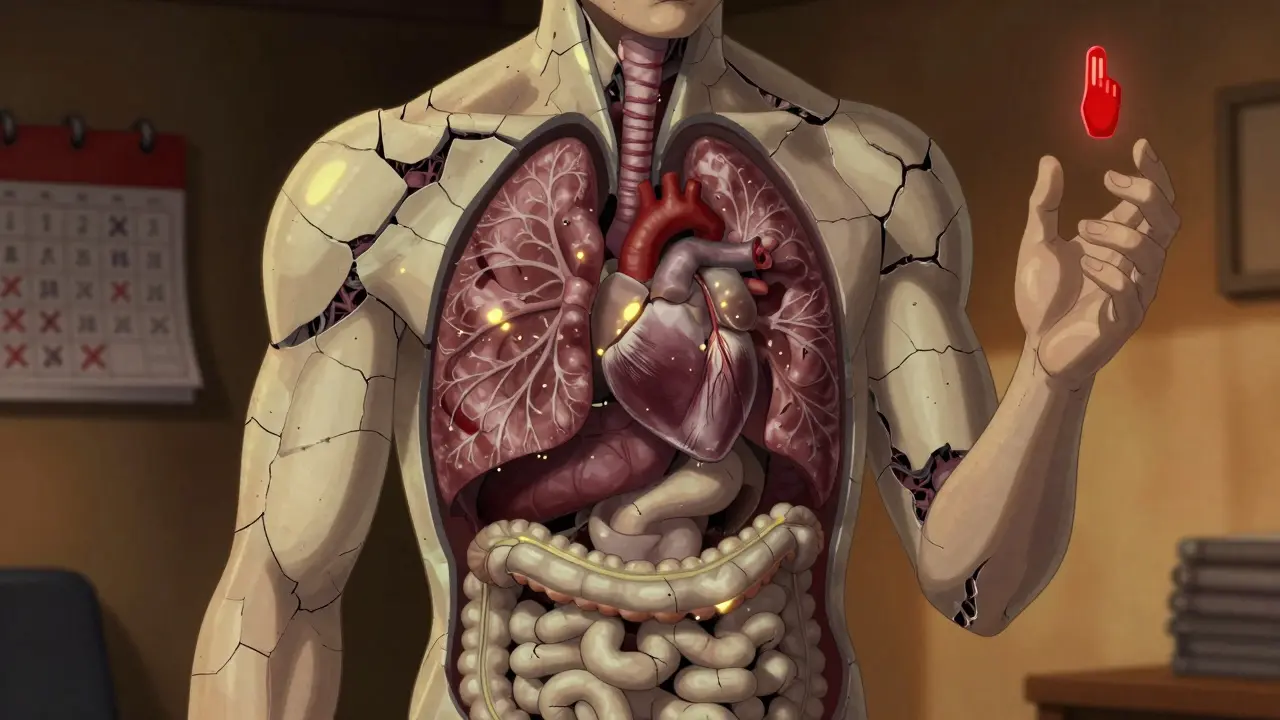
What Happens When It Progresses
There are two main forms of systemic scleroderma: limited and diffuse.
Limited cutaneous (formerly CREST syndrome) progresses slowly. Skin thickening usually stops at the fingers and face. It’s less aggressive, but it carries a high risk of pulmonary arterial hypertension. People with this form often live 10-20 years after diagnosis. The 10-year survival rate is 75-85%.
Diffuse cutaneous hits fast. Skin thickens quickly over the arms, legs, and torso. It’s more likely to damage lungs and kidneys. The first 3-5 years are critical. If the lungs get scarred or blood pressure in the lungs spikes, survival drops. The 10-year survival rate here is only 55-70%.
And then there’s the gut. Ninety percent of patients have trouble with digestion. Food moves slowly. Acid reflux is constant. Some can’t absorb nutrients. Others get bacterial overgrowth that causes bloating and diarrhea. Medications help, but diet changes are just as important.
Treatment Today-And Why It’s Not Enough
There are no FDA-approved drugs that cure scleroderma. Everything used is borrowed from other diseases.
For Raynaud’s, calcium channel blockers like nifedipine open up blood vessels. For lung fibrosis, tocilizumab became the first drug specifically approved for scleroderma-related lung disease in 2021. For pulmonary hypertension, drugs like bosentan and riociguat help lower pressure in the lungs.
But here’s the hard truth: less than 30% of patients with diffuse scleroderma see real improvement in skin hardness after a year of immunosuppressants. Most treatments only slow things down. They don’t reverse damage.
That’s why multidisciplinary care matters. A single rheumatologist can’t handle all the complications. You need a pulmonologist for lung scans, a cardiologist for heart monitoring, a gastroenterologist for digestion, and a wound care specialist for those painful digital ulcers that form on fingertips. These ulcers affect 60% of patients and can take weeks to heal.
Life With Scleroderma-The Real Daily Struggles
It’s not just medical. It’s daily life.
78% of patients say they can’t button shirts, open jars, or hold utensils because their fingers are stiff. 65% use adaptive tools-grip aids, electric can openers, zipper pulls. 70% are too tired to work full-time. 82% have constant reflux. 45% have severe acid damage to their esophagus.
One woman in Auckland told her doctor she stopped cooking because she couldn’t hold a knife. Another stopped gardening because her hands wouldn’t open. A man in his 40s had to quit his job as a mechanic because he couldn’t turn a wrench.
And the emotional toll? Depression and anxiety are common. Isolation is too. Many feel like no one understands what it’s like to wake up with hands that don’t work, or to have your lungs feel like they’re wrapped in plastic.

What’s New in Research
Hope is growing. In 2024, the Scleroderma Research Foundation committed $15 million to new fibrosis-targeting drugs. Over 47 clinical trials are active, testing everything from B-cell blockers to tyrosine kinase inhibitors.
One breakthrough: autologous stem cell transplants. In the 2023 SCOT trial, half of patients with severe diffuse scleroderma saw their skin soften by 50% after 4.5 years. It’s risky-some patients had serious side effects-but for those with rapidly worsening disease, it’s a game-changer.
Another: a new blood marker called CXCL4 shows up before symptoms. If validated, it could let doctors diagnose scleroderma before organs are damaged.
Telemedicine is helping too. Stanford’s virtual care program cut hospital visits by 32% for rural patients. That’s huge in places like New Zealand, where specialists are far apart.
How to Get the Best Care
Only 35% of U.S. patients see a scleroderma specialist. In New Zealand, the number is probably lower. But care at a dedicated center-like Johns Hopkins, Stanford, or the University of Michigan-makes a difference. Patients there report 68% better symptom control than those seeing general rheumatologists.
Here’s what you need: a rheumatologist who specializes in scleroderma, plus regular check-ins with a pulmonologist, cardiologist, and GI specialist. Track your symptoms: skin thickness, Raynaud’s episodes, breathing, digestion. Use a journal. Take photos of your fingers. Bring logs to every appointment.
And don’t wait for the perfect doctor. If your current doctor doesn’t know about anti-Scl-70 or digital ulcers, ask for a referral. You deserve care that understands this disease.
What’s Next?
Scleroderma is getting more attention. More funding. More research. More awareness. But for now, it’s still a disease that sneaks up on people. If you’ve had Raynaud’s for years, and your skin feels tighter, or your fingers won’t bend, or you’re constantly tired-don’t brush it off. Talk to a rheumatologist. Ask for the right tests.
There’s no cure yet. But with early detection and the right team, you can live longer. Better. And with more control over your life than ever before.





Alvin Bregman
January 14, 2026 AT 15:57Man i had raynauds for years thought it was just cold fingers till my knuckles got stiff like rubber bands
Andrew Freeman
January 15, 2026 AT 20:01so basically its just your body thinking its in a war and keeps building bunkers out of collagen
says haze
January 16, 2026 AT 02:16It's not merely a medical anomaly-it's a metaphysical betrayal of the body's innate intelligence. The immune system, once a guardian, becomes the architect of its own prison. Collagen, the very scaffold of tissue, is perverted into a prison wall. This isn't disease-it's ontological failure. The body doesn't malfunction; it mutates into its own opposite. And yet, we treat it like a glitch in a software update, not a collapse of biological covenant.
Modern medicine, with its pharmacological Band-Aids and diagnostic delay rituals, fails to confront the existential horror of scleroderma. We quantify fibrosis with CT scans, but we refuse to acknowledge the grief of a woman who can no longer hold her child's hand. We name antibodies like trophies, but we don't name the silence that follows when someone says, 'I can't button my shirt anymore.' This isn't rheumatology. This is elegy written in scar tissue.
Dylan Livingston
January 17, 2026 AT 17:13Oh wow, another ‘rare disease’ that somehow gets more attention than actual public health crises. I mean, sure, 1 in 3 have heart complications-but how many people are dying from TikTok addiction? Or from being told to ‘drink more water’ for chronic pain? At least scleroderma has fancy acronyms and NIH grants. Meanwhile, my cousin with fibromyalgia gets told she’s ‘just stressed.’
And let’s not forget the ‘women are 4x more likely’ bit. Is that because biology is cursed, or because we only start looking when it’s a ‘woman’s issue’? I’m sure if men got stiff fingers from cold, it’d be called ‘elite athlete syndrome’ and get $5 billion in funding by Tuesday.
Sarah -Jane Vincent
January 18, 2026 AT 11:27Did you know the CDC knows about this but hides it because Big Pharma doesn’t want a cure? They’re all invested in lifelong immunosuppressants. That’s why stem cell transplants are ‘risky’-they’re not approved because they’d put drug companies out of business. Look at the timeline: 2021 tocilizumab approval, then boom-$15 million in research? Coincidence? I think not. CXCL4 marker? Probably been known since 2018. They’re waiting for the patent window to close.
And don’t get me started on ‘specialists.’ You think Johns Hopkins cares? They’re billing codes and insurance networks. Real treatment? It’s cold baths, magnesium, and avoiding gluten. I cured my cousin’s Raynaud’s with Himalayan salt and a sauna. No drugs. No biologics. Just primal wisdom they don’t want you to know.
Henry Sy
January 19, 2026 AT 06:51imagine your skin turning into shrink wrap and your lungs feeling like theyre stuffed with concrete
and the worst part? your fingers look fine but you cant even hold a damn coffee mug without dropping it
they call it rare but i know three people who got diagnosed after being told they were ‘just old’ or ‘anxious’
its like your body forgets how to stop fixing itself
Anna Hunger
January 20, 2026 AT 22:45Thank you for this comprehensive and meticulously researched overview. The emphasis on early detection, multidisciplinary care, and patient-driven documentation is not merely advisable-it is essential. I have reviewed the clinical guidelines from the American College of Rheumatology and the European League Against Rheumatism, and your summary aligns precisely with the latest consensus. The data on pulmonary arterial hypertension and digital ulcers is particularly vital for primary care providers who may lack exposure to this condition.
I urge all readers to maintain symptom journals, photograph skin changes monthly, and request referral to a scleroderma center if Raynaud’s persists beyond three years. Early intervention alters trajectory. This is not advocacy-it is evidence-based medicine.
Jason Yan
January 21, 2026 AT 23:48It’s wild how something so internal-this silent overproduction of collagen-ends up reshaping entire lives. You don’t just lose mobility; you lose the small things. The way your fingers used to brush your partner’s hair. The way you could open a jar without crying. The way you didn’t have to think about breathing.
But here’s the quiet miracle: people with scleroderma are learning to adapt. Not just with gadgets, but with creativity. One woman I read about started painting with her feet. Another learned to type with her chin. The body adapts, even when it’s betraying you. Maybe that’s the real lesson here-not just about disease, but about what resilience looks like when it has no fanfare.
We talk about curing it, and we should. But let’s not forget to celebrate the people who are living with it, fully, fiercely, even when the world doesn’t see them.
shiv singh
January 23, 2026 AT 13:39you think this is bad? wait till you see what the government is doing to the immune system with vaccines and 5G. they want you weak so you stay dependent on drugs. scleroderma? its a manufactured disease to sell more pills. i saw a guy in india with the same symptoms and he never took one pill-he just ate turmeric and prayed. cured in 3 months. they dont want you to know that
Robert Way
January 25, 2026 AT 11:19so if you have raynauds and your skin feels tight you should go to a specialist? what if you dont have insurance? or live in a town with no rheumatologist? what then? just die?
Sarah Triphahn
January 27, 2026 AT 09:22people like you think this is tragic but honestly? you chose this. your body’s just reacting to your stress, your bad diet, your negative thoughts. you’re literally thinking yourself into fibrosis. stop being a victim and start taking responsibility. meditate. detox. stop eating gluten. problem solved.
Vicky Zhang
January 27, 2026 AT 16:12I just want to say-this post made me cry. Not because I have it, but because I know someone who does. My best friend, Lisa. She’s 34. She can’t hold her dog anymore. She cries every night because she can’t hug her mom. But she still laughs. She still makes art with adaptive tools. She still shows up. I don’t know how she does it. But I’m so proud of her. And if you’re reading this and you have scleroderma-you’re not alone. You’re a warrior. I see you.
Allison Deming
January 28, 2026 AT 20:43It is imperative to recognize that the narrative surrounding scleroderma often romanticizes suffering while neglecting systemic failures in healthcare accessibility. The assertion that ‘early detection saves lives’ is only valid when early detection is economically and geographically attainable. In rural America, where specialists are scarce and telemedicine is underfunded, this becomes a privilege, not a right. The $15 million in research funding is commendable-but it is a drop in the ocean compared to the $2.3 billion spent annually on cosmetic dermatology. The disparity is not accidental. It is structural. We must demand equitable access-not just awareness.